Болезнь Коновалова-Вильсона называется так в честь докторов, впервые давших детальную характеристику заболевания, предложивших не только критерии диагноза, но и возможные варианты терапии заболевания.
Под этим сочетанием двух фамилий следует понимать наследственное нарушение обмена микроэлемента меди, которое в случае отсутствия необходимой терапии приводит к тяжелым поражениям всех тканей печени и головного мозга, вплоть до летального исхода.
При своевременно начатой терапии человек с гепатоцеребральной дистрофией (она же болезнь Коновалова-Вильсона) ничем не отличается от здорового. Наиболее часто это заболевание впервые проявляется в школьном и подростковом возрасте, значительно реже – у взрослого человека.
Причины появления и общие особенности патологии
Непосредственные причины развития гепатоцеребральной дистрофии в настоящее время не известны.
Морфологической основой этого заболевания является мутация гена, который располагается в 13й хромосоме человека. Это провоцирует значительные нарушения обмена микроэлемента меди в человеческом организме: этот микроэлемент не связывается с соответствующим белком (церулоплазмин), плохо выводится наружу с током желчи и, соответственно, накапливается в тканях печени и прочих внутренних органов.
Такое избыточное содержание меди во всех тканях приводит к формированию разнообразных воспалительных и дистрофических процессов.
Болезнь Коновалова-Вильсона имеет следующие особенности:
- для развития заболевания необходимо наличие в генотипе у человека двух мутаций выше указанного гена, для этого оба родителя должны иметь либо такой же вариант генотипа, либо является носителями комбинации мутации и нормального гена (так называемая гетерозигота);
- болезнь Коновалова-Вильсона наиболее актуальна для малых народов и этнических групп, где приняты близкородственные брачные союзы;
- частота встречаемости этого наследственного заболевания несколько выше в европейских странах, чем в государствах Юго-Восточной Азии;
- болезнь Коновалова-Вильсона в одинаковой степени актуальна у ребенка мужского и женского пола;
- наиболее часто первые симптомы заболевания появляются у детей в возрасте от 10 до 18 лет, быстрота нарастания клинической симптоматики определяется индивидуальными особенностями и формой болезни.
Клиническая классификация
Гепатолентикулярная дегенерация подразделяется современными педиатрами на 5 клинических форм в соответствии преимущественным поражением определенного органа (головного мозга или печени) и соответствующих клинических признаков болезни. Среди них выделяют:
- экстрапирамидно-корковая обусловлена выраженными изменениями тканей головного мозга, проявляется развитием тяжелых параличей и слабоумием; продолжительность без необходимого лечения не более 6-8 лет;
- дрожательно-ригидная форма встречается наиболее часто, длится в пределах 5-6 лет, клинические признаки соответствуют названию;
- дрожательная форма впервые проявляется в подростковом возрасте, прогрессирует очень медленно, постепенно изменяется психика пациента, отмечаются судорожные припадки;
- ригидно-аритмогиперкинетическая наиболее актуальна для пациентов дошкольного возраста, проявляется разнообразными нарушениями психики и двигательных функций, прогрессирует очень быстро;
- брюшная форма характеризуется преимущественным поражением печени.
Клинические проявления выше описанных форм не отличаются у детей различного возраста, сходными являются общие принципы лечения и диагностики.
Клиническая симптоматика
Симптомы заболевания определяются преимущественным поражением печени, головного мозга или почек. В таком порядке и следует их рассматривать, подчеркивая важность сочетанного поражения внутренних органов.
Для поражения печени при этом заболевании типичны:
- увеличение размеров органа и периодические болезненные ощущения в правом подреберье тянущего и ноющего характера;
- желтушно-коричневый оттенок кожи, интенсивность которого нарастает, а также эпизод младенческой желтухи у новорожденного;
- склонность к повышенной кровоточивости (обусловлена нарушением синтеза факторов свертывания в печени), что проявляется кровотечениями из носа и десен, длительным
- кровотечением при небольших бытовых травмах;
на поздних стадиях болезни, при сформировавшемся циррозе отмечаются все характерные признаки – асцит, расширение венозной сети на коже живота, сосудистые звездочки, - красные (печеночные) ладони.
Поражение ЦНС (головного мозга) при гепатоцеребральной дистрофии характеризуется значительным разнообразием, а именно:
- у ребенка могут наблюдаться судорожные припадки, напоминающие эпилептические;
- нередко обращают на себя задержка умственного и эмоционального развития, проблемы с обучением в обычной среднестатистической школе;
- при дрожательных формах отмечаются практически постоянные дрожания конечностей, которые усиливаются при эмоциональном или физическом напряжении, значительно затрудняют даже самые простые бытовые действия;
- возникают приступы депрессивного настроения и немотивированного возбуждения (дурашливости);
- по мере прогрессирования болезни прогрессируют нарушения координации движений и речи, значительно снижается уровень интеллекта.
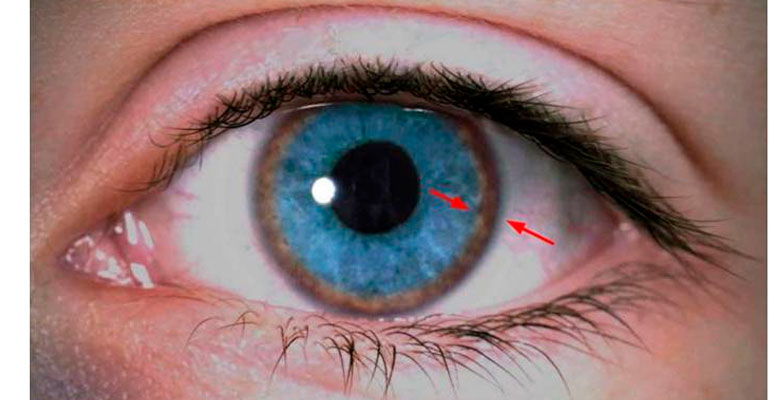
Одним из характерных внешних признаков этого наследственного патологического процесса является – буровато-зеленый кружок вокруг радужной оболочки – кольцо Кайзера-Флейшера. Нередко отмечается задержка развития вторичных половых признаков у девочек и мальчиков.
Особенности диагностики заболевания
Гепатоцеребральная дистрофия может быть окончательно диагностирована только на основании специфических исследований. К ним относят:
- изучение в крови маленького пациента уровня содержания меди (значительно снижена);
- исследование уровня выведения меди с суточной мочой (повышается);
- изучение уровня содержания специфического, связывающего микроэлемент медь в крови, белкового соединения церулоплазмина (снижается);
- исследование гена ATP7B для выявления его мутаций.
В процессе детальной диагностики могут потребоваться многочисленные биохимические и инструментальные исследования для комплексной оценки здоровья маленького пациента.
Общие принципы лечения
Терапия гепатоцеребральной дистрофии основана на двух главных моментах:
- применение препаратов с действующим веществом пеницилламин, которые связывают избыток меди и нормализуют процессы ее метаболизма;
- строгое соблюдение диеты №5 (исключение жирных, жареных, острых блюд), а также ингредиентов, которые содержат большое количество меди (бобовые, орехи, шоколад, кофе).
Продолжительное (пожизненное) использование этих составляющих (под контролем доктора) позволяет достичь обратного развития клинических признаков и длительной ремиссии гепатоцеребральной дистрофии.
Прогноз лечения
Благоприятный при своевременном и постоянном специфическом лечении. В настоящее время считается целесообразным начинать терапию пенилламином еще до начала клинической симптоматики болезни, то есть при минимальных нарушениях обмена микроэлемента.
Специалисты комментируют гепатоцеребральную дистрофию как тяжелое наследственное заболевание тканей печени и головного мозга, но вполне управляемое и обратимое под воздействием специфической терапии. Чем раньше начато лечение, тем более успешным оно будет.